IM 7 Family
This is a case study that was analyzed in one of our articles where we implemented FrustraEvo as a web server to study protein families stability and functional patterns: Parra and Freiberger et al BiorXiv 2023.
In the following link you can find a FrustraEvo reproduction of this example:
FrustraEvo job results
Biological Interpretation:
The algorithm to localize local energetic frustration was first introduced in 2007 with an accompanying work where frustration patterns, and their consequences for the folding mechanisms of Im7, a protein with known 3D structure, were presented.
Im7 is an 87 amino acids single domain protein that inhibits the bacterial toxicity of its cognate protein, colicin E7. It has been shown that the residues that are located at the Im7 surface, on the interaction site with colicin E7, become highly frustrated when Im7 is alone. This frustration in turn has been shown to be correlated with an increased flexibility in that region. Moreover, this frustration is known to be linked to the appearance of an on-pathway folding intermediate state that is stabilized by non-native interactions. This high frustration in the native state of Im7 is released when the Im7-colicin E7 complex is formed. This is a clear example of the tradeoff that exists between stability and function in proteins. Residues that are used by evolution to perform specific molecular functions are often not favorable to local stability, making those proteins susceptible to detrimental phenomena such as aggregation or misfolding. Evolution has however found different strategies to minimize such risks, for example, through the use of chaperones. The Spy chaperone was shown to selectively recognise and interact with the highly frustrated residues within the interacting interface in Im7. An unfrustrated state can subsequently be achieved by the interaction of Im7 with colicin E7, which releases it from the chaperone.
Structural information has been a limitation to study frustration on a large scale. The evolutionary context of proteins can be used to define regions that are constrained to change even when they are energetically detrimental, i.e. are highly frustrated. The recent advances in efficient structure prediction techniques, allow us to overcome such limitations and unblocked a whole new set of possibilities to perform high-throughput frustration analysis. In what follows we analyze the conservation of frustration levels within a set of Im7 homologs to better understand the evolutionary constraints of those proteins. In Fig. A the Multiple Sequence Frustration Alignment (MSFA) shows the distribution of both amino acid identities and frustration states for every aligned residue across the family members. The information content of the sequence (SeqIC) and frustration (FrustIC) are shown in the form of logos to visualize such conservation (Fig. B). Conservation of frustration levels is heterogeneous at different loci in the MSFA. Some residues show high variability both in sequence and in frustration e.g. position 17 that can be found as minimally frustrated, neutral or highly frustrated in different proteins of the family. This is reflected in the frustration logo (Fig. B) with no high FrustIC values for those positions. Some other regions can be variable in their amino acid identities but still be conserved in their frustration state, e.g. position 19 that although varying among the L, M, V or I amino acids stays in a minimally frustrated state. This is reflected with that position having a not so high SeqIC but a maximum FrustIC. Several positions composed of hydrophobic residues follow this pattern, highlighting their importance for the fold stability as being part of the hydrophobic core (green, tall bars in Fig. B). There are 3 residues that are highly frustrated and conserved in most members of the family (red, tall bars in Fig. B). Two of these residues are Y55 and Y56 that belong to the interacting region of Im7 with colicin E7 and Spy. In addition, W75 is also frustrated and conserved although it is not located at the protein-protein interaction site. W75, however, is reported to establish non-native interactions that are present at the intermediate state, possibly stabilizing it. If the intermediate state is an undesired consequence of the functional requirements associated to the binding residues used to bind colicin E7 why is there a residue that is detrimental to the local energetics in the native state that stabilizes it? Could it be possible that the intermediate itself has been positively selected? Notably, according to the MSFA, there is one protein in the dataset (WP199883497.1) that has that residue deleted. It would be of interest to perform MD simulations to assess whether this protein has such a folding intermediate state or not.
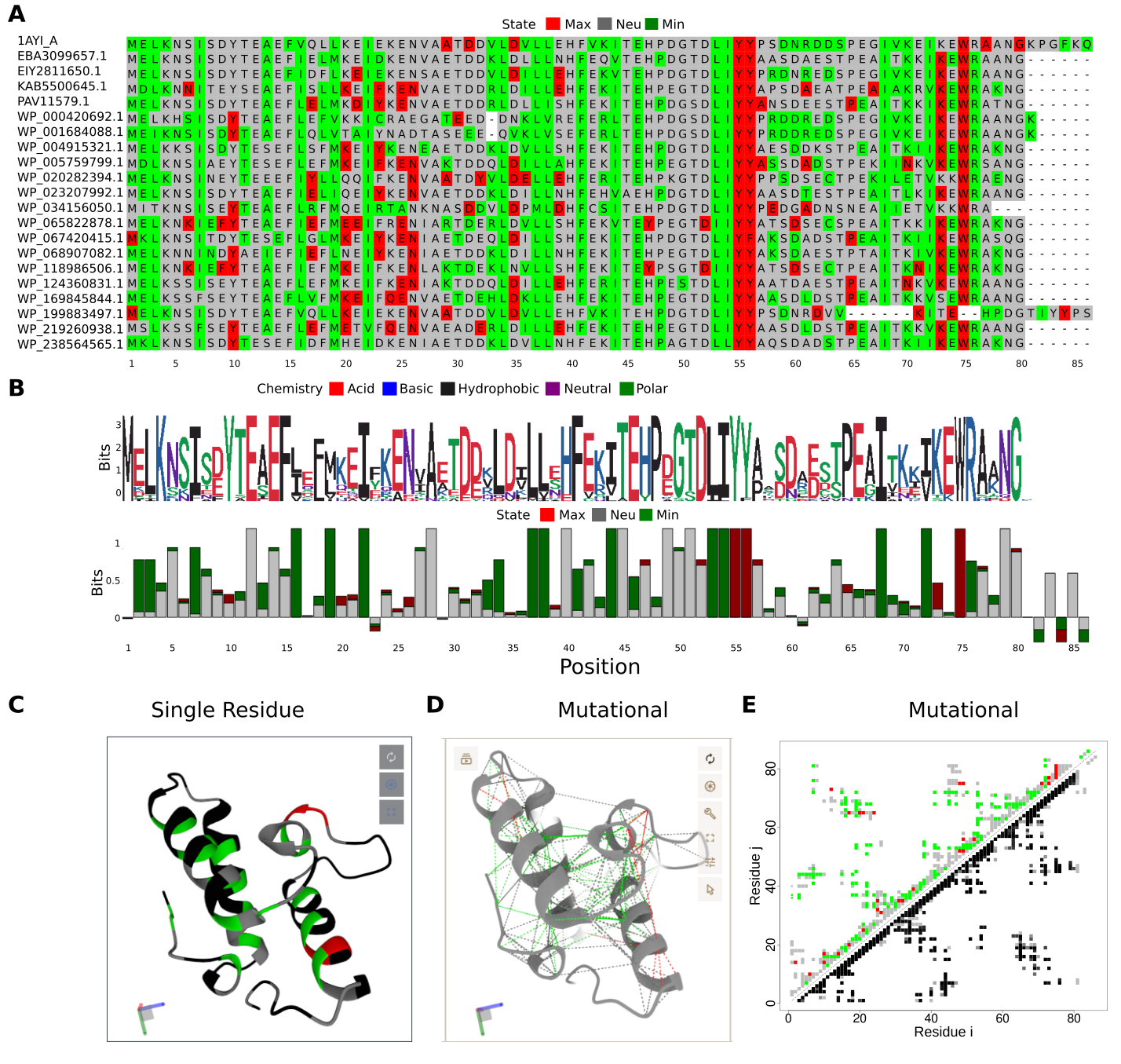
Different visualizations as produced by the FrustraEvo web server using PdbID=1AYI as the reference structure. A) Multiple Sequence Frustration Alignment (MSFA): Each residue in the MSA used as input is coloured according to its frustration state, mapped from the corresponding structure. B) Sequence and frustration logos: sequence and frustration conservation are visually represented as logos where SeqIC and FrustIC are displayed for each position. The individual contribution of each symbol to the total IC in each case is represented by the relative size of it to the size of the bars. C) Protein structure colored according to FrustIC based on SRFI as visualized on the web server. Residues with FrustIC >= 0.5 are coloured according to their most informative frustration value and residues with FrustIC < 0.5 are coloured in black. D) Protein structure of 1AYI with its contacts colored according to FrustIC values based on the mutational mode (only contacts with FrustIC >= 0.5 are shown) as it can be visualized in Pymol locally on the user’s computer. E) Contact maps: The FrustIC values, obtained from the contacts mode using the mutational FI are represented. In the upper diagonal matrix each point represents a contact between residues i and j where its color is assigned according to its most informative frustration state. In the lower diagonal matrix each point represents the same contacts as in the upper diagonal matrix but coloured in shadows of gray, representing the proportion of structures in the dataset that contain that contact between residues i and j.